Texto universitario

_____________________________
Módulo 5. Química organometálica y organometálica de metales alcalinotérreos pesados
5.1 Introducción
Durante la última década, la química organometálica y de metales alcalinotérreos pesados ??ha pasado de la oscuridad a un área emocionante y de rápido desarrollo de la química. Entre los desarrollos más recientes se encuentra el uso de los compuestos en catálisis o en transformaciones orgánicas, lo que indica aplicaciones que no se creían posibles hace solo 20 años. Con estos desarrollos, los compuestos de metales organoalcalinotérreos están emergiendo como una rama reconocida de la química, con un potencial significativo en una amplia gama de aplicaciones que incluyen síntesis y catálisis, química de polímeros y aplicaciones de materiales[1]. La clave para el surgimiento de la química orgánica de metales y organometálicos de metales alcalinotérreos es el examen y desarrollo de protocolos sintéticos que permiten la preparación y el aislamiento de compuestos objetivo que luego pueden emplearse en una variedad de aplicaciones. Como tal, el examen detallado de metodologías sintéticas establecidas y la exploración de nuevas rutas de reacción han sido fundamentales para el crecimiento de la química de metales alcalinotérreos. Las dificultades encontradas se basan en la alta reactividad de los compuestos resultantes, lo que ha llevado a severas restricciones de metodologías sintéticas viables. Los metales más ligeros, el berilio y el magnesio, con sus radios pequeños, muestran relaciones carga/tamaño relativamente altas, lo que coincide con la capacidad de polarización de enlaces y la inducción de covalencia de enlaces. Como tal, entre los metales alcalinotérreos, estos elementos muestran enlaces metal-carbono con un mayor carácter covalente que proporciona especies relativamente estables. Debido a la toxicidad del berilio y sus compuestos[2], se ha realizado poco trabajo, pero los compuestos que contienen magnesio han sido más intensamente estudiado como lo demuestra el uso establecido de reactivos de Grignard y diorganomagnesio en aplicaciones sintéticas[3]. Por el contrario, el radio más grande, el potencial redox fuertemente negativo, combinado con alta hidro y oxofilicidad, alto carácter electropositivo y la falta de orbitales d vacíos y energéticamente accesibles son responsables de las dificultades encontradas cuando se trabaja con los metales alcalinotérreos pesados y sus compuestos.
Los metales reaccionan fácilmente con el oxígeno o el agua formando óxidos o hidróxidos; los compuestos resultantes son muy sensibles a la hidrólisis. Además, el enlace metal-ligando suele ser bastante débil, lo que da lugar a una labilidad significativa debido a las grandes diferencias de electronegatividad; los enlaces metal-ligando son en gran medida electrostáticos, lo que contribuye aún más a la labilidad de los compuestos objetivo. Sus grandes radios iónicos[4] (![]() ) no solo promueven los enlaces de ligando metálico, sino que también son responsables de la baja solubilidad, ya que a menudo se logra la saturación estérica de los metales por agregación que requiere el uso de codisolventes polares para romper los agregados. Exacerbando el problema de solubilidad, muchos compuestos de metales organoalcalinotérreos reaccionan fácilmente con solventes etéreos bajo la química de escisión de éter, lo que limita aún más las variables sintéticas. Este problema a menudo se puede superar reduciendo la temperatura; por lo tanto, muchas reacciones se llevan a cabo a -40 C o menos.
) no solo promueven los enlaces de ligando metálico, sino que también son responsables de la baja solubilidad, ya que a menudo se logra la saturación estérica de los metales por agregación que requiere el uso de codisolventes polares para romper los agregados. Exacerbando el problema de solubilidad, muchos compuestos de metales organoalcalinotérreos reaccionan fácilmente con solventes etéreos bajo la química de escisión de éter, lo que limita aún más las variables sintéticas. Este problema a menudo se puede superar reduciendo la temperatura; por lo tanto, muchas reacciones se llevan a cabo a -40 C o menos.
Como se muestra en este manuscrito, la exploración en profundidad de vías sintéticas establecidas y el desarrollo de nuevas metodologías dan lugar a una química emocionante con muchas oportunidades. Los métodos clásicos que involucran magnesio se han modificado para extenderse hacia los congéneres más pesados, y se ha examinado la química de otros sistemas metálicos para ofrecer nuevas estrategias.
Por ejemplo, la relación carga/tamaño cercana para:
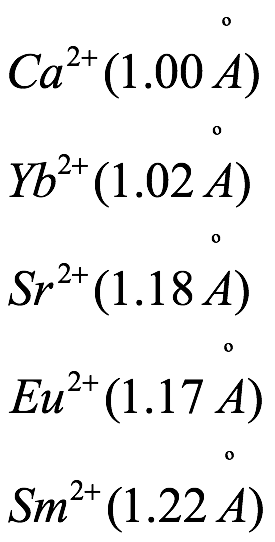
hace que la exploración paralela de su química sea emocionante.
A pesar de mostrar similitudes estructurales, los potenciales redox más negativos para los metales alcalinotérreos 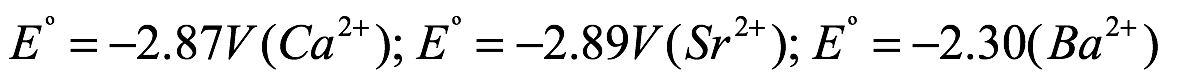 los hacen más reactivos que sus equivalentes lantánidos (
los hacen más reactivos que sus equivalentes lantánidos (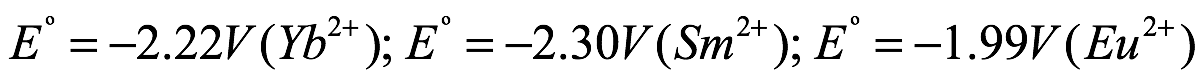 .
.
Comencemos con algunas preguntas simples: ¿qué es la química organometálica? ¿Qué, después de estudiar química organometálica, sabremos del mundo que no sabíamos antes? ¿Por qué vale la pena estudiar el tema? ¿Y qué tipo de problemas pretende abordar el tema? El propósito de esta publicación es dar las mejores respuestas que conozco actualmente a estas preguntas. El objetivo de este manuscrito es doble: (1) ayudarnos a motivarnos a medida que avanzamos (es decir, ¡recordarnos constantemente que todo esto tiene un sentido!); y (2) para ilustrar los tipos de problemas que podremos abordar usando conceptos.
En pocas palabras, la química organometálica (OM) es el estudio de compuestos que contienen enlaces metal-carbono y reacciones que involucran. El enlace metal-carbono puede ser transitorio o temporal, pero si existe durante una reacción o en un compuesto de interés, estamos de lleno en el dominio de la química organometálica. A pesar de la importancia denotativa del enlace M-C, los enlaces entre los metales y los otros elementos comunes de la química orgánica también aparecen en la química OM: los enlaces metal-nitrógeno, metal-oxígeno, metal-halógeno e incluso metal-hidrógeno juegan un papel. Los metales cubren una amplia franja de la tabla periódica e incluyen los metales alcalinos (grupo 1), los metales alcalinotérreos (grupo 2), los metales de transición (grupos 3-12), los metales del grupo principal (grupos 13-15, "bajo las escaleras”), y los lantánidos y actínidos. Nos centraremos principalmente en el comportamiento de los metales de transición, llamados así porque cubren la transición entre los elementos electropositivos del grupo 2 y los elementos del grupo principal más ricos en electrones.
¿Por qué vale la pena estudiar el tema? Bueno, se trata principalmente de flexibilidad sintética. Hay una razón por la que el "organo" ocupa el primer lugar en la "química organometálica": el objetivo suele ser la creación de nuevos enlaces en los compuestos orgánicos. Los metales tienden a estar solo para el viaje (aunque su influencia, obviamente, es esencial). Y el hecho es que puedes hacer cosas con la química organometálica que no puedes hacer usando la química orgánica directa. Caso puntual:
 06.49.22.png)
La venerable reacción de Suzuki... ¡impensable sin paladio!
El establecimiento del enlace entre los anillos de fenilo a través de un medio que no sea pura suerte parece impensable para el químico orgánico, pero es natural para la organometálica con paladio. El bromobenceno parece un electrófilo potencial en el carbono que contiene bromo, y si está familiarizado con la hidroboración, es posible que vea al ácido fenilborónico como un nucleófilo potencial en el carbono que contiene boro. ¡El paladio catalítico hace que todo suceda! La química organometálica está llena de estas transformaciones alucinantes y puede expandir considerablemente la caja de herramientas sintéticas del químico orgánico.
Para agregar otro motivo a la mezcla para el no especialista (o el químico que rechaza la síntesis), la química organometálica está llena de historias intrigantes de investigación y descubrimiento científico. Explorar cómo los investigadores toman una nueva reacción organometálica de "oh hermosa” a un fuerte poder predictivo es instructivo para cualquier persona interesada en "cómo funciona la ciencia", en un sentido práctico.
Ahí lo tiene, una breve introducción a la química organometálica y por qué vale la pena estudiarla. Por supuesto, usaremos el resto del espacio para describir completamente qué es realmente la química organometálica... pero es útil tener estos motivos en mente mientras estudia. (La química organometálica se encarga del estudio, la síntesis y la reactividad de los compuestos organometálicos, aquellos compuestos químicos que poseen al menos un enlace entre un átomo de carbono de un ligando orgánico y un átomo metálico. En este contexto, el término metal se puede definir utilizando una escala de electronegatividad, asignando la palabra metal a aquel elemento que presenta un carácter menos electronegativo que el carbono. Desde este punto de vista, se designan como metales a elementos conocidos como metaloides, tal como el silicio[5]).
5.2 Reactivos de Grignard
El descubrimiento innovador de la preparación de haluros de organomagnesio mediante el tratamiento de magnesio metálico con un haluro orgánico en presencia de éter dietílico de Victor Grignard en 1900 proporcionó un acceso sintético fácil e innovador. Debido a la versatilidad y aplicabilidad de los reactivos en una amplia gama de aplicaciones sintéticas, los reactivos de Grignard a base de magnesio se encuentran entre las especies organometálicas del grupo principal más ampliamente estudiadas.
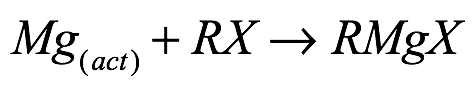
Donde act=activado; X=F, Cl, Br, I; R=alkyl, aryl[6]. Ec. (1)
Química análoga que involucra a los congéneres de metales más pesados ??[Ec. (2)] ha tenido un éxito limitado hasta hace muy poco tiempo. La mayor reactividad de los compuestos objetivo hace que su preparación sea más desafiante que la de los derivados de magnesio, ya que la combinación de un metal relativamente poco reactivo con los compuestos objetivo altamente reactivos plantea un desafío sintético único. Complicaciones adicionales implican la alta tendencia de los reactivos hacia la escisión del éter, lo que limita significativamente las condiciones de reacción.
El primer reactivo de Grignard de metal alcalinotérreo pesado, EtCaI, fue informado por Beckmann; sin embargo, no se dispone de datos analíticos detallados. El trabajo actual indica que la especie de Beckmann era, de hecho[7], . Los intentos de Gilman dieron como resultado hallazgos similares[8]. Del mismo modo, varios otros informaron resultados similares, pero solo recientemente se han puesto a disposición reactivos de Grignard de base alcalinotérrea pesada que están completamente caracterizados[9]. De manera similar a la preparación de magnesio de Grignard, la síntesis del calcio y, más recientemente, la de los análogos de estroncio y bario dependen en gran medida de la pureza del metal de origen, que se puede lograr mediante la destilación de los metales[10]. La destilación también elimina la capa de óxido, proporcionando así un medio significativo de activación de metales. Sin embargo, la destilación no da como resultado el aumento del área superficial del metal, un factor importante en la reducción del tiempo de reacción[11]. Los métodos efectivos de activación incluyen la reducción de los yoduros de metales alcalinotérreos con potasio metálico o naftalida de litio, la preparación de amalgamas para mejorar la superficie mediante la adición de mercurio, métodos de co-condensación[12], y solvatación de los metales en amoníaco anhidro, líquido o gaseoso. Más comúnmente, el metal alcalinotérreo se disuelve en amoníaco, con una rápida eliminación del solvente para evitar la formación de la amida (M(NH2)2), lo que da como resultado un polvo metálico altamente reactivo. Como la tendencia hacia la formación de amidas aumenta con el radio del metal, esta metodología se ha utilizado con mayor éxito para el calcio; sin embargo, para el estroncio y el bario, la formación de M(NH2)2 es difícil de evitar. Además, la presencia de perlas de vidrio en el matraz puede aumentar el área superficial total del metal activado. Este método se ha utilizado con éxito para el calcio. Como resultado, se informaron varios derivados del arilo cálcico (Fig. 1), mientras que solo se identificaron una especie de bario y una de estroncio.
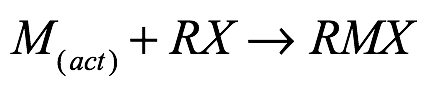
Donde M=Ca, Sr, Ba; R=aryl; X=Cl, Br, I. Ec. (2)
 18.10.43.png)
Fig. 1 Estructura cristalina de MesCaI(thf)4 (Mes o 2,4,6-trimetilfenilo). Átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados
 18.13.00.png)
Esquema 1. Una ruta de descomposición generalizada para complejos de haluro de arilo cálcico. El protón ácido del coligando THF se extrae, lo que produce benceno, seguido de la liberación de eteno y la formación de un etenolato de arilo cálcico. El mecanismo anterior se propone en base a las observaciones de los experimentos de RMN y está de acuerdo con otras rutas establecidas de descomposición por escisión del éter.
De acuerdo con la alta reactividad de los haluros de aril calcio, las reacciones de escisión del éter son frecuentes. De manera análoga a los reactivos de Grignard a base de magnesio, existe una fuerte dependencia de los solventes, obteniéndose mejores resultados en THF (tetrahidrofurano) y tiempos de reacción más largos en éter dietílico. No se produce ninguna reacción en los hidrocarburos alifáticos y aromáticos. Las reacciones de escisión del éter generalmente se suprimen con tiempos de reacción cortos y mantienen las temperaturas de reacción en o por debajo de -40º C. Sin embargo, la escisión del éter ocurre fácilmente de acuerdo con el mecanismo común que se muestra en el Esquema 1.
Existe una diferencia significativa en la reactividad de los haluros de arilo y alquilo, siendo los yoduros los más reactivos. Como resultado, el uso de yodoarenos ha tenido mucho éxito. Un ligero aumento de la temperatura pone a disposición los derivados del bromo; sin embargo, los fluoro y cloroarenos no muestran reactividad. De manera análoga a los reactivos de Grignard de magnesio, los derivados de calcio correspondientes también exhiben equilibrios de Schlenk dependientes de la temperatura y el solvente donde se pueden identificar los derivados de calcio de dihaluro y diarilo. A pesar del creciente éxito en la preparación de análogos de calcio de Grignard, los compuestos permanecen altamente reactivos y requieren el control más estricto de la calidad del material de partida junto con la exclusión más estricta de oxígeno y humedad. Sin embargo, la alta reactividad de los compuestos también los hace atractivos en aplicaciones sintéticas, específicamente debido al alto pKa de los derivados del benceno (pKa ~ 43). Esta ruta se ha utilizado para sintetizar ciclopentadienuros, fosfuros, naftilos y especies de calcio de baja valencia. Como lo demostraron impresionantemente Westerhausen et al., la estabilidad de los reactivos de calcio de Grignard depende críticamente del número de coordinación de los metales. La sustitución de tres donantes en el PhCaI(thf)4 de seis coordenadas por dos co-ligandos DME (dimetoxietano) produce el PhCaI(dme)2 de siete coordenadas que da como resultado un aumento de 30º C en la estabilidad del compuesto. Además, la estabilidad de los compuestos organometálicos objetivo también se puede aumentar aumentando la demanda estérica de los ligandos de arilo empleando haluros de arilo sustituidos (MesBr, pTolBr, 2,6-dimetoxifenilBr, 2,6-pTolBr, naftilBr) como se muestra para Mes2Ca(thf)3 (Fig. 2). Sin embargo, la sustitución en la posición orto puede dar lugar a reordenamientos y, en algunos casos, a un acoplamiento de tipo Wurtz.
 18.16.54.png)
Fig. 2 Estructura cristalina de Mes2Ca(thf)3 sintetizada mediante el uso de Grignards pesados en THF. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados
5.3 Eliminación u organoeliminación de hidrocarburos
La organoeliminación ha sido el caballo de batalla para la preparación de una amplia gama de especies a base de magnesio debido a la disponibilidad comercial o al fácil acceso al reactivo de diorganomagnesio [Ec. (3)]. Específicamente, el Bu2Mg comercial (una mezcla estadística entre la especie secundaria y la de n-butilo) proporciona un fácil acceso a los bencilatos, ciclopentadienuros, indenuros, acetiluros, amidas, alcóxidos, arilóxidos, fluoroalcóxidos, silóxidos, tiolatos, fosfanuros, selenolatos, telurolatos y arseniuros en alto rendimiento y pureza. La diferencia significativa en la acidez entre el n/segBuH resultante (pKa ~ 53) y la mayoría de los ligandos impulsa la reacción, mientras que la naturaleza volátil del subproducto butano proporciona un procesamiento sencillo. Los aductos donantes se obtienen fácilmente mediante la adición de co-ligando, preferiblemente después de que haya tenido lugar la reacción (Fig. 3).
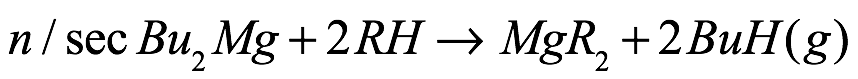 Ec. 3
Ec. 3
donde RH = benceno, ciclopentadieno, indeno, acetileno, amina, alcohol, fenol, fluoroalcohol, siloxano, tiol, fosfano, selenol, telurol , arsano.
Se ha demostrado que la organoeliminación depende del solvente y del ligando, y la presencia de solventes polares conduce con frecuencia a reacciones incompletas con retención de uno de los dos restos alquilo que dan como resultado especies heterolépticas (Fig. 4). Como resultado, las reacciones se realizan preferentemente en disolventes no polares como hexano o tolueno. Sin embargo, el uso de disolventes que no donan normalmente da como resultado la agregación bajo la formación de dímeros o agregados superiores para sistemas de ligandos menos estéricamente exigentes, mientras que se pueden obtener compuestos monoméricos libres de donantes para ligandos estéricamente más exigentes (Fig. 5). En el caso de ligandos muy exigentes estéricamente, se requiere reflujo durante la noche para la formación del producto.
 18.26.02.png)
Fig. 3 Estructura cristalina de Mg[N(SiMe3)(Mes)]2(hmpa)2 (HMPA o triamida hexametilfosfórica) sintetizada mediante organoeliminación en hexano. Los átomos de hidrógeno y los grupos metilo de HMPA se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
 18.27.28.png)
Fig. 4 Estructura del heteroléptico [(t-Bu)Mg(thf)]2 sintetizado mediante organoeliminación en THF. Los átomos de hidrógeno de todos los grupos metilo se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados
Si bien proporciona un fácil acceso para las especies de magnesio, esta ruta es menos directa para los metales alcalinotérreos más pesados ??debido a las limitaciones de los reactivos de dialquilo o arilo adecuados. Sin embargo, la preparación reciente de reactivos de dibencilo de los metales alcalinotérreos pesados ??M(PhCH2)2 (M =Ca–Ba) (Fig. 6) y su reacción con ligandos ácidos bajo liberación de tolueno han proporcionado una poderosa estrategia sintética [Ec. (4)].
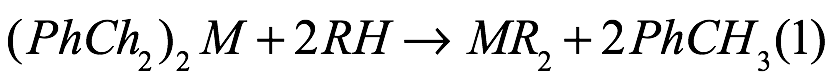 Ec. 4
Ec. 4
donde M = Ca, Sr, Ba; RH = di- y trifenilmetano, acetileno, ciclopentadieno, amina.
 18.36.30.png)
Fig. 5 Estructura de Mg[N (SiMe3)(Dipp)]2 (Dipp o 2,6-diisopropilfenilo) sintetizado mediante organoeliminación en hexano. Las interacciones de Mg … C (π) se muestran como líneas discontinuas. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados
 18.36.36.png)
Fig. 6 Estructura cristalina de Ca(PhCH2)2(thf)4 sintetizada mediante metátesis de sal en THF. Los átomos de hidrógeno no bencílicos se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
Dado que el pKa del tolueno es bastante alto (pKa ~ 41), los reactivos de dibencilo permiten la metalación de muchos sistemas de ligandos. Además, la formación de tolueno como producto secundario facilita el procesamiento y, por lo tanto, el aislamiento de las moléculas diana. Las limitaciones de la ruta se refieren a la baja solubilidad de los reactivos de dibencilo en hidrocarburos que requieren el uso de THF para lograr condiciones de reacción homogéneas. Sin embargo, la naturaleza altamente reactiva de los compuestos de dibencilo da como resultado la escisión del éter si las temperaturas no se mantienen por debajo de los -40 C.
Varias rutas de reacción proporcionan acceso sintético a los reactivos de dibencilo, históricamente relacionados con la transmetalación redox que involucra la reacción de un espejo de bario con dibencilmercurio. Una ruta más conveniente involucra la reacción del M[N(SiMe3)2]2(thf)2 (M = Sr–Ba) de fácil acceso con el reactivo de intercambio de metales Li(PhCH2)tmeda (TMEDA, N,N ,N’,N’-tetrametil-1,2-etilendiamina).
 18.44.05.png)
Fig. 8 Estructura cristalina de Ba(Ph2CH)2(18-corona-6) sintetizada mediante organoeliminación en THF. Las interacciones Ba???C(π) se muestran como líneas discontinuas. Los átomos de hidrógeno no bencílicos se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
Estas reacciones se pueden realizar en Et2O; sin embargo, se obtienen productos de reacción más limpios cuando la reacción se realiza en tolueno. Sin embargo, la química similar para el análogo de calcio sigue siendo difícil, como se muestra con el aislamiento del calciato de bencilo heterobimetálico obtenido por el tratamiento de bencilitio con Ca[N(SiMe3)2]2(thf)2 en presencia de TMEDA (Fig. 7). El calciato también se puede obtener por tratamiento de Ca(PhCH2)2(thf)4 con dos equivalentes de Li(PhCH2)tmeda (Esquema 2). Una comparación de la reactividad entre el Ca(PhCH2)2(thf)4 homometálico y el calciado muestra una reactividad significativamente mayor para las especies de calciato heterobimetálico, un resultado que está de acuerdo con las tendencias de reactividad de otros complejos “ate” organometálicos alcalinotérreos. La eliminación de tolueno se ha utilizado para producir una familia de complejos de di- y trifenilmetánidos alcalinotérreos altamente reactivos (Fig. 8), que muestran un rango de modos de asociación de iones dependiendo de los co-ligandos. Otros ejemplos incluyen acetiluros, ciclopentadienuros y amidas.
 18.49.07.png)
Esquema 2 Vías de reacción hacia (tmeda)2Li2Ca(PhCH2)4
5.4 Transaminación
El desarrollo de bis(bis(trimetilsilil)amidas de metales alcalinotérreos pesados, M[N(SiMe3)2]2(thf)2, ha sido fundamental en el uso de la ruta de transaminación como una ruta sintética viable y confiable hacia la preparación de compuestos de metales alcalinotérreos. Las amidas, M[N(SiMe3)2]2(thf)2, se pueden preparar mediante metátesis de sal que implica el tratamiento de los haluros metálicos con amida de metal alcalino (Ca-Ba). Las amidas de metales más pesados ??(Sr, Ba) también se pueden obtener por metalación directa en amoníaco anhidro líquido (Sr y Ba).
Recientemente, la transmetalación redox/intercambio de ligandos (RTLE) en presencia de Ph3Bi como reactivo de transmetalación ha proporcionado las amidas de metales alcalinotérreos pesados ??con alto rendimiento y pureza del metal en una conveniente reacción en un solo recipiente, una reacción que se analizará con más detalle. Además de la facilidad de preparación de las bis(bis(trimetilsilil)amidas, su estabilidad y solubilidad en un La amplia gama de disolventes polares y no polares los convierte en materiales de partida muy atractivos. La transaminación implica la reacción de las bis(bis(trimetilsilil)amidas con un sustrato ácido (pKa < 30) [Ec. (5)] con liberación de amina libre, HN (SiMe3)2 (pKa ~ 30). El bajo punto de ebullición de la amina resultante permite su fácil eliminación al vacío y, por lo tanto, facilita el procesamiento. Como se muestra en [Ec. (5)], la reacción se basa en un equilibrio; como tal, los rendimientos del producto pueden ser aumenta por la eliminación rápida de la amina liberada
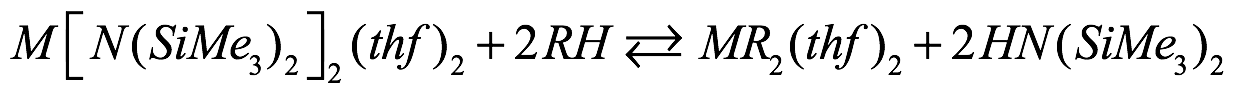 Ec. (5)
Ec. (5)
donde M = Mg, Ca, Sr, Ba; RH = benceno, di- y trifenilmetano, ciclopentadieno, acetileno, pirazol, amina, guanidina, alcohol, fluoroalcohol, tiol, fosfano, selenol, telurol, arsano.
Sin embargo, la principal limitación para las reacciones de transaminación es el pKa del hexametildisilazano (pKa ~ 30), ya que solo sustratos con un pKa más bajo que ese del HN (SiMe3)2. Los ejemplos representativos de productos obtenidos incluyen bencilatos, di- y trifenilmetánidos, cíclope ntadienidas, acetilidas, pirazolatos, amidas, guanidinatos, alcóxidos, fluoroalcóxidos (Fig. 9), tiolatos, fosfanuros, selenolatos, telurolatos y arseniuros. Por otro lado, los ligandos altamente ácidos pueden promover la escisión del enlace N-Si en el resto trimetilsililo, lo que da como resultado la formación de amina primaria [Eq. (6)]; esta reacción es especialmente frecuente en presencia de un exceso de ácido. Una segunda protonación produce NH3. La escisión del enlace N-Si se puede suprimir mediante la adición lenta y diluida de un ligando ácido.
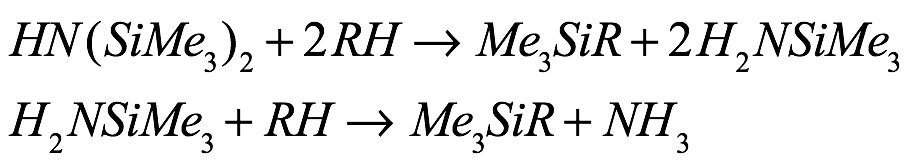 Ec. (6)
Ec. (6)
donde RH = alcano, areno, silano.
Además, se ha informado que las bis(bis(trimetilsilil) amidas de metales alcalinotérreos inician la química de escisión del éter, como se muestra con la apertura del anillo de los éteres corona o THF.
 05.47.27.png)
Fig. 9 Estructura cristalina de Sr[OC(CF3)3]2(thf)4 sintetizada mediante transaminación en THF. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
5.5 Metátesis de sal
Metátesis de sal [Ec. (7 )] es una ruta bien establecida en la síntesis de una variedad de alquilos de metales alcalinotérreos, alilos, bencilatos, ciclopentadienuros, pentadienilos, fluorenilos, indenilos, amida, β-dicetiminatos, guanidinatos, alcóxidos, arilóxidos, silanidas, tiolatos, fosfanidas, selenolatos y germanidas. Sin embargo, la ruta rara vez se ha utilizado para la síntesis de más complejos de metales de alquilo y arilo reactivos, en gran parte debido a problemas relacionados con la química de escisión del éter, ya que la metátesis generalmente requiere la presencia de un solvente etéreo.La metátesis de sal es un procedimiento de dos pasos; el paso inicial involucra ves metalación del ligando. Hay diferentes agentes de metalización disponibles; convenientemente, se pueden usar hidruros de metales alcalinos, ya que liberan gas hidrógeno limpiamente bajo la formación cuantitativa de los derivados de metales alcalinos. Como los hidruros muestran una solubilidad limitada, incluso en THF, los agentes metalantes alternativos que se usan con frecuencia son las bis(trimetilsilil)amidas de metales alcalinos. Lo más frecuente es que se utilicen derivados del potasio (véase más adelante); como tal, la siguiente narrativa a continuación se enfoca específicamente en esos. La formación de los derivados de potasio va seguida de su adición in situ a los haluros de metales alcalinotérreos poco solubles en THF. Entre los haluros, los fluoruros son los menos solubles, mientras que los yoduros presentan la mayor solubilidad. Para los metales más pesados, solo se pueden lograr condiciones de reacción homogéneas si se emplean los yoduros junto con THF como disolvente. Además, la reacción depende críticamente de haluros de alta calidad.
 05.44.20.png) Ec. (7)
Ec. (7)
donde M = Be–Ba; A = Li–K; X = Cl, Br, I; RH = alcano, aleno, benceno, ciclopentadieno, pentadieno, fluoreno, indeno, amina, β-dicetimina, guanidina, alcohol, fenol, silano, tiol, fosfano, selenol, germano.
La reacción es impulsada por la precipitación del haluro de metal alcalino. En THF, se ha demostrado que el KI normalmente precipita limpiamente y proporciona los mejores resultados. Por lo tanto, la combinación de reactivos de potasio (ver arriba) con los yoduros se ha empleado ampliamente en esta ruta. Las limitaciones de esta ruta de reacción incluyen la solubilidad limitada de los yoduros de metales alcalinotérreos y los derivados de potasio, lo que requiere la necesidad de soluciones etéreas. Esta necesidad de disolventes polares puede dar lugar a reacciones de escisión del éter. Si bien la eliminación de sales se ha empleado para la preparación de los metalocenos, sigue siendo una ruta desafiante hacia otros grupos de organometálicos debido a la necesidad de preparar precursores de potasio altamente reactivos. Como se mencionó anteriormente, la metátesis de sal se lleva a cabo normalmente en THF debido a la mayor solubilidad del material de partida. Eaborn et al. demuestran muy bien los problemas asociados con la metátesis de sal en la síntesis de Ca[C(SiMe3)3]2 (Fig. 10). La exposición de este compuesto al éter dietílico conduce a la escisión inmediata del éter bajo la formación de Ca(OEt)2. Como resultado, la reacción de metátesis de sal se realizó en benceno. Debido a las limitaciones de solubilidad, el tiempo de reacción se extendió a 48 h, lo que resultó en mayores rendimientos. Ejemplos de compuestos organometálicos preparados por metátesis de sal en THF incluyen M[C(SiMe3)3]2(thf)n (M = Ca, n = 2;M = Sr, Ba, n = 3), Ca(CH2Ph )2(thf)4 (Fig. 6) y más recientemente Ca[C(SiHMe2)3]2(thf)2.
 05.52.40.png)
Fig. 10 Estructura cristalina de Ca(C(SiMe3)3)2 sintetizada mediante metátesis de sal en benceno. Las interacciones Ca???C(π) se muestran como líneas discontinuas. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
5.6 Metalación directa
La preparación de organometálicos de metales alcalinotérreos pesados ??que implica la reacción del metal con un sustrato de ligando ácido sigue siendo una estrategia sintética muy sencilla, que generalmente requiere una superficie metálica libre de óxido. Para sistemas de ligandos altamente ácidos, las condiciones de reflujo son suficientes. La metalación directa también se puede emplear para el magnesio, como lo demuestra la preparación de derivados de antraceno de magnesio en presencia de THF; sin embargo, se requiere polvo metálico finamente disperso. Sin embargo, los sistemas menos ácidos se benefician de la activación del metal. Esto se puede lograr a través de varias vías, incluido el uso de metal de alta pureza y/o una variedad de métodos de activación que incluyen la reducción de los yoduros de metales alcalinotérreos con metal de potasio o naftalida de litio, preparación de amalgamas para mejorar la superficie mediante la adición de mercurio, así como métodos de co-condensación, También se exploraron ampliamente las reacciones de los metales en amoníaco anhidro, líquido o gaseoso, como se analiza en detalle a continuación. Recientemente, la química del estado sólido suave, que involucra el tratamiento de los metales en presencia del ligando y un agente fundente en ausencia de un solvente, ha tenido mucho éxito en la preparación de varios arilóxidos de metales alcalinotérreos pesados, incluido el heterobimetálico especies [AM(ODpp)3] (ODpp o 2,6-difenilfenóxido) (A = Na, K; M =Ca–Ba) o [Li2Ba(ODpp)4].
5.6.1 Metalación directa a través de la activación del NH3(l) anhidro
La fácil solubilidad de los metales alcalinotérreos pesados ??en amoníaco líquido anhidro, bajo la formación de electrones solvatados, proporciona un acceso conveniente a una forma altamente reactiva de los metales [Ec. (8)]. A medida que aumenta la reactividad de los metales al descender en el grupo, las reacciones que involucran al bario ocurren más rápidamente, mientras que se requieren condiciones de reflujo para el estroncio y el calcio (pe NH3 = -33 C). Coincidiendo con una mayor reactividad, la propensión a formar el M(NH2)2 insoluble es mucho más frecuente para el estroncio y el bario. M(NH2)2 puede reaccionar con sustratos ácidos bajo la formación de NH3.
Los metales disueltos se tratan con dos equivalentes de ligando ácido que da como resultado la eliminación de hidrógeno, bajo un aislamiento limpio de los compuestos objetivo. La presencia de co-ligando polar promueve la reacción, como lo demuestra la síntesis de bis(bis(trimetilsilil)amidas, donde la presencia de THF promueve mayores rendimientos. Los contratiempos de la ruta incluyen el trabajo con gases condensados (pb de NH3 =- 33 C) y la purificación de amoníaco (condensación sobre sodio), ya que el NH3 gaseoso comercial no es anhidro
 05.56.44.png) Ec. (8)
Ec. (8)
donde M = Ca, Sr, Ba, RH = ciclopentadieno, fluoreno, amina, pirazol, antraceno, alcohol, fenol, fluoroalcohol, tiol, silano, siloxano, selenol.
La metalización directa a través de la activación con amoníaco anhidro ha sido se utiliza para preparar con éxito ciclopentadienidas, fluorenilos, amidas, pirazolatos (Fig. 11), antracenidas, alcóxidos, arilóxidos, fluoroalcóxidos, tiolatos, silanidas, silóxidos, selenolatos y un metanuro raro, Ba[(PhCH (C5H3N-2))]2 (diglima) (thf)]. Aductos de amoníaco escasamente solubles de la con frecuencia se observan complejos metálicos, pero la eliminación del amoníaco se puede lograr calentando para formar el complejo objetivo.
 06.01.00.png)
Fig. 11 Estructura cristalina de [{Ba(Ph2pz)2(tmeda)}2]?TMEDA sintetizada mediante metalación directa mediante activación de amoníaco en tolueno. Las interacciones Ba???C(π) se muestran como líneas discontinuas. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
5.6.2 Metalación directa en condiciones de estado sólido leves
La metalización directa en condiciones de estado sólido leves evita la posible ruptura química del éter al eludir la exposición de derivados de metales alcalinotérreos pesados ??a solventes etéreos. La combinación de muestras de metales puros con ligandos ácidos, a menudo en presencia de un agente fundente en condiciones de reacción suaves (250 C) en tubos Carius (vidrio de paredes gruesas) sellados y evacuados, permite la preparación de derivados alcalinotérreos con altos rendimientos y pureza [Ec. (9) ]. La ausencia de co-ligandos y disolventes polares da como resultado una fácil preparación y cristalización de compuestos sin co-ligandos (Fig. 12). Estas reacciones se pueden monitorear fácilmente por la evolución del gas dihidrógeno de la superficie del metal.
 06.01.43.png) Ec. (9)
Ec. (9)
donde M =Ca, Sr, Ba; RH = pirazol, fenol.
La adición de mercurio puede facilitar la formación de productos para el calcio y los lantánidos menos reactivos a través de la formación de amalgamas metálicas, pero generalmente no es necesario para los metales alcalinotérreos más pesados, en consonancia con sus propiedades más negativas potenciales redox. En algunos casos, es necesaria la presencia de un medio de reacción inerte o agente fundente que tenga un punto de fusión bajo, especialmente si el ligando no se derretirá a las temperaturas de reacción. Un ejemplo de un agente fundente eficaz es el 1,2,4,5-tetrametilbenceno o el 1,2,5-tri-terc-butilbenceno (Mes*H). En algunos casos, el propio ligando demuestra ser un medio de reacción adecuado. El enfriamiento lento de las mezclas de reacción produce con frecuencia cristales con calidad de rayos X de compuestos homolépticos libres de coligandos. El aislamiento de los compuestos objetivo es sencillo: el lavado de las mezclas de reacción con hexano eliminará el ligando y el agente fundente que no hayan reaccionado. La extracción de productos brutos en disolventes polares o en presencia de co-ligandos proporciona los respectivos productos que contienen co-ligandos. Esta ruta de reacción ha tenido éxito en la preparación de varias especies de pirazolatos alcalinotérreos, así como arilóxidos de metales alcalinotérreos. Es importante destacar que las técnicas han tenido éxito en la preparación de compuestos heterobimetálicos alcalino-alcalino, alcalino-lantánido y alcalino-alcalino que, en presencia de un disolvente donante, se descomponen en los compuestos originales homometálicos. Debido a la ausencia de bases de Lewis, los compuestos diana sin co-ligando exhiben diferentes motivos estructurales que los homólogos que contienen co-ligando. Los compuestos sin coligandos son especialmente atractivos para la investigación de interacciones débiles no covalentes, incluidas M???C(π) e interacciones agósticas que con frecuencia proporcionan saturación estérica para los grandes centros de metales alcalinotérreos (Fig. 13).
 06.10.42.png)
Fig. 12 Estructura de [Sr4(tBu2pz)8] sintetizado mediante metalación directa en condiciones sin disolventes. Las interacciones Sr???C(π) se muestran como líneas discontinuas. Los grupos metilo en los átomos de hidrógeno y del sustituyente terc-butilo se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
Si bien la activación de metales en condiciones suaves de estado sólido proporciona una ruta de reacción directa, se deben abordar algunas limitaciones y problemas de seguridad. La acidez y, en última instancia, la estabilidad térmica de los ligandos presentan la principal preocupación en esta ruta de reacción. Dado que se necesitan temperaturas superiores a 150 C para un reacción suave, la descomposición parcial del ligando es típicamente una preocupación. Además, el control cuidadoso de la escala de reacción es crítico ya que los gases se generan en un sistema cerrado.
5.7 Transmetalación redox
La transmetalación redox es la ruta clásica para preparar compuestos organometálicos alcalinotérreos. Esta ruta aprovecha la naturaleza electropositiva de los metales alcalinotérreos haciéndolos reaccionar con complejos organometálicos de mercurio, estaño o talio de sacrificio [Ec. (10)]. Este método proporciona buenos rendimientos de producto en cualquiera de los solventes polares, pero también se observan rendimientos aceptables en solventes aromáticos o de hidrocarburo, al tiempo que proporciona una purificación directa del producto que implica la eliminación del reactivo sin reaccionar y el producto metálico por filtración, seguido de la cristalización del compuesto objetivo a partir de un disolvente adecuado.
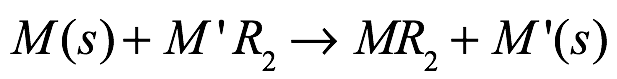 Ec. (10)
Ec. (10)
donde M = Ca, Sr, Ba; M’ = Hg, Sn; R= bis(trimetilsilil)amida; M’ = Tl; R=pirazolato.
A este efecto, los organomercuriales se han utilizado ampliamente como agentes transmetalantes para producir compuestos de magnesio y organoberilio libres de coligandos puros. Los mercuriales también se han utilizado para la preparación de bis(bis(trimetilsilil)amidas. Más recientemente, la química de transmetalación basada en estaño permitió la preparación de las amidas, mientras que los pirazolatos de talio se emplearon en la síntesis de los correspondientes derivados alcalinotérreos [Ca(Ph2pz)2(thf)4] y [Sr(Ph2pz)2(dme)2] (Ph2pz o difenilpirazolato). La limitación de esta ruta es la toxicidad inherente de los agentes de transmetalación redox.
 06.12.56.png)
Fig. 13 Estructura cristalina de [BaSr(Odpp)4] sintetizada mediante metalación directa en condiciones sin disolventes. Las interacciones M???C(π) se muestran como líneas discontinuas. Tenga en cuenta la disminución del número de interacciones M???C(π) en el bario más grande debido a la presencia de un ligando de arilóxido terminal. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
5.7.1 Transmetalación redox/intercambio de ligandos
La transmetalación redox/intercambio de ligandos (RTLE) proporciona una variante de la transmetalación redox. Esta ruta normalmente utiliza piezas o limaduras de metales alcalinotérreos y un arilo (C6H5) o, con menos frecuencia, pentafluoroarilo (C6F5) mercurial como agente transmetalante con la formación in situ de un intermedio organometálico alcalinotérreo reactivo.Este intermedio no es estable en las condiciones de reacción y es seguido inmediatamente por el intercambio de ligandos que implica un sustrato prótico en formato ion del compuesto objetivo y liberación de benceno o pentafluorobenceno [Eq. (11)]. Las reacciones de esta naturaleza se realizan típicamente como una reacción de un solo recipiente en presencia de un disolvente polar. Esta metodología está muy bien establecida en la química de los lantanoides (usando mercuriales como agente de transmetalación redox) y se ha utilizado para la preparación de una variedad de compuestos organometálicos alcalinotérreos que incluyen ciclopentadienuro, pirazolatos, formamidinatos y arilóxidos.
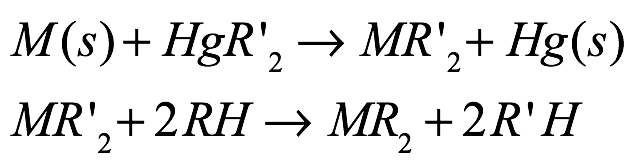 Ec. (11)
Ec. (11)
donde M = Ca, Sr, Ba; R’ = C6F5, C6H5; R = ciclopentadienuro, pirazolato, formamidinato, arilóxido.
Intrínsecamente crucial para la reacción es la presencia de un ligando con un pKa más bajo que el benceno resultante (pKa ~ 43) o pentafluorobenceno (pKa ~ 26). Además, los tiempos de reacción dependen del tamaño del metal como consecuencia de potenciales redox más negativos al descender en el grupo de metales alcalinotérreos. Se han realizado avances recientes en esta estrategia sintética reemplazando el organomercurial tóxico por el agente de transmetalación Ph3Bi [Eq. (12)]]. Además de ser menos tóxico que el clásico organomercurial [LD50 Ph3Bi: 180 g kg 1 (perro, oral); LD50 Ph2Hg: 50–400 mg kg 1 (rata, oral)], Ph3Bi ofrece varias ventajas: está disponible comercialmente, es económico y estable al aire y la humedad, y permite un fácil procesamiento de los compuestos objetivo mientras proporcionando buenos rendimientos de producto. Como la reacción depende de la diferencia de potenciales redox entre los dos metales, el mercurio, con su potencial redox positivo, funciona muy bien (Eº = 0,851 V (Hg)). Reemplazar el mercurio con el bismuto menos positivo (Eº = 0,308 V(Bi)) limita así la ruta de reacción a los metales con los potenciales redox más negativos, como los metales alcalinotérreos pesados ??(Eº = 2,87 V(Ca2+) E = 2,89 V (Sr2+), Eº = 2,90 V (Ba2+)). Los metales de tierras raras con sus potenciales redox menos negativos (Eº = 2,22 V(Yb2+); Eº = 2,30 V(Sm2+); Eº = 1,99 V(Eu2+)) no funcionan bien con Ph3Bi.
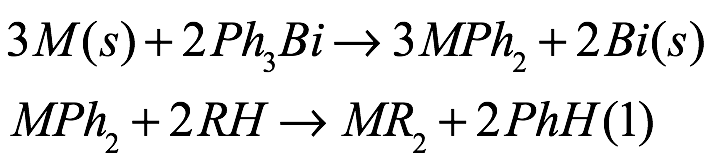 Ec. (12)
Ec. (12)
donde M = Ca, Sr, Ba; RH = ciclopentadieno, amina, pirazol, fenol.
De forma análoga al intercambio de ligandos de transmetalación redox en presencia de organomercuriales, son necesarios ligandos con pKa más bajo que el benceno resultante (pKa ~ 43). Los tiempos de reacción dependen del tamaño del metal, las reacciones que involucran bario ocurren en tiempos más rápidos que los metales más livianos. La optimización de las condiciones de reacción, incluido el uso de limaduras metálicas y la sonicación, ha permitido la preparación de varias amidas de metales alcalinotérreos pesados ? (Fig. 14), ciclopentadienuros y arilóxidos.
 06.25.07.png)
Fig. 14 Estructura cristalina de Ba[N(Mes)(SiMe3)]2(thf)3 sintetizada mediante transmetalación redox/intercambio de ligandos (RTLE). Las interacciones Ba???C(π) se muestran como líneas discontinuas. Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoides sombreados.
5.8 Intercambio de metales
Las reacciones de intercambio de metales se han usado en la preparación de compuestos organometálicos alcalinos pesados ??por reacción de alcóxido o arilóxido de metal alcalino con un reactivo de organolitio bajo precipitación de alcóxido/arilóxido de litio. Para estas reacciones, la elección cuidadosa del ligando permite la separación de los dos productos de reacción sólidos. Se han informado dos variantes de esta reacción para organometálicos de metales alcalinotérreos: reacción de alcóxido alcalinotérreo pesado con bencilitio [Eq. (13)] y tratamiento de las bis(bis(trimetilsilil)amidas alcalinotérreas con un reactivo de litio [Ec. (14)].
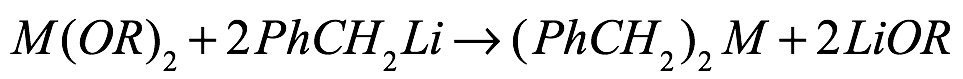 Ec. (13)
Ec. (13)
donde M = Ba ; R = 2,4,6-tri-tercbutilfenilo
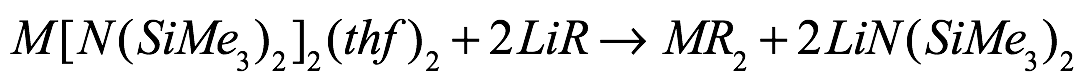 Ec. (14)
Ec. (14)
donde M = Ca, Sr, Ba R = bencilo, di-, trifenilmetánido.
Como se ve para metales alcalinos, la separación de los dos productos de reacción sólidos es el principal inconveniente de esta ruta. Además, los terc-butóxidos de metales alcalinotérreos son difíciles de preparar en forma pura, ya que las reacciones de escisión del éter inician con frecuencia óxidos o hidróxidos que contienen (Westerhausen, Private Communication). Alternativamente, se pueden usar las correspondientes amidas de fácil acceso. Estas también ayudarán en el aislamiento de los productos. Debido a la naturaleza soluble de la amida de litio resultante, la separación de los productos es más fácil, ya que los productos normalmente precipitan El uso de bis(bis(trimetilsilil)amidas alcalinotérreas junto con lit bencílico hium ha proporcionado un camino para la preparación con altos rendimientos de organometálicos alcalinotérreos en forma de bencilatos que son los materiales de partida necesarios para las reacciones de organoeliminación que conducen a di- y trifenilmetánidos (ver arriba) (Fig. 15).
 06.34.28.png)
Fig. 15 Estructura cristalina de [Ca(N(SiMe3)2)(18-corona-6)][CPh3] (CPh3 o trifenilmetánido). Los átomos de hidrógeno se han omitido para mayor claridad. Todos los átomos que no son de carbono se muestran como elipsoide sombreado.
5.9 Conclusiones
El rápido desarrollo de la química organometálica y organometálica alcalinotérrea ha sido posible gracias al examen y, a menudo, a las modificaciones de las estrategias sintéticas establecidas, además de la exploración de metodologías alternativas. La comprensión del impacto de las variables de reacción, incluida la temperatura de reacción y la elección del disolvente, ha sido fundamental en la síntesis de nuevas especies de metales alcalinotérreos. Estos desarrollos seguirán avanzando en la química de los metales alcalinotérreos y proporcionarán las herramientas necesarias para la preparación de compuestos con aplicaciones sintéticas y de materiales.
Referencias
[1] Chirik, Paul & Ritter, Stephen. (2021). 40 Years of Organometallics. Organometallics. 40. 4035-4040.
DOI:10.1021/acs.organomet.1c00667.
[2] Soni, P.L. & Soni, Vandna. (2021). Organometallics.
DOI:10.1201/9781003183426-16.
[3] Alexander, Jacob. (2022). Not just Grignards: New developments in the organometallic chemistry of the heavy alkali and alkaline earth metals. Chemistry - Dissertations and Theses.
[4] https://journals.iucr.org/a/services/about.html
[5] https://es.wikipedia.org/wiki/Qu%C3%ADmica_organometálica
[6] https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/alkyl-aryl-ketone
[7] Gasik, Mikhail & Dashevskii, Viktor & Bizhanov, Aitber. (2020). Alkaline Earth Metal Ferroalloys.
DOI:0.1007/978-3-030-57502-1_12.
[8] Eisch, John. (2002). Henry Gilman: American Pioneer in the Rise of Organometallic Chemistry in Modern Science and Technology†. Cheminform. 34.
DOI:10.1021/om0109408.
[9] Huang, Wenliang & Diaconescu, Paula. (2022). Organometallic Chemistry of Lanthanides.
DOI:10.1142/9781800610163_0006.
[10] Ammon, HL & Wheeler, GL & Agranat, I. (1973). Tetrahedron, 29, 2695 (1973). Tetrahedron. 29. 2695.
[11] Proctor, S.G.. (1976). A new process for americium purification. J Less-Common Metals. Journal of The Less Common Metals. 44. 195-199.
DOI:10.1016/0022-5088(76)90131-4.
[12] Seyferth, Dietmar. (2004). Uranocene. The First Member of a New Class of Organometallic Derivatives of the f Elements. Organometallics. 23.
DOI:10.1021/om0400705.
Autores:
Eduardo Ochoa Hernández
Nicolás Zamudio Hernández
Abraham Zamudio Durán
Lizbeth Guadalupe Villalon Magallan
Mónica Rico Reyes
Pedro Gallegos Facio
Gerardo Sánchez Fernández
Rogelio Ochoa Barragán